iELM workflow
iELM
identifying human SLiM-mediated interaction
Background
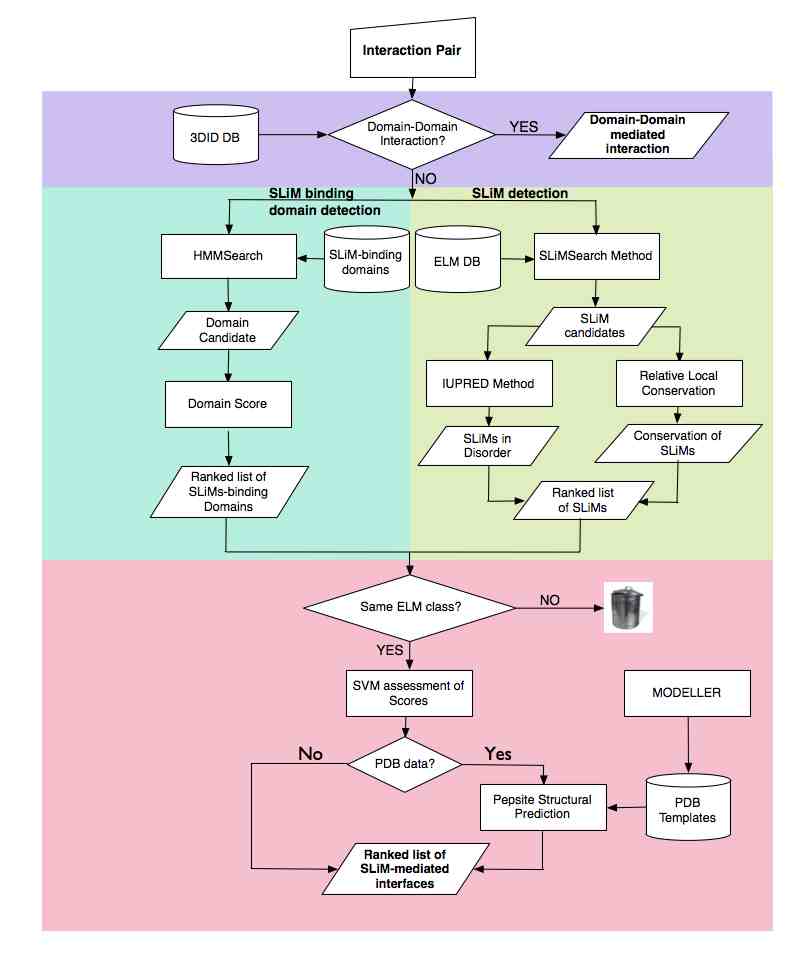
iELM aims to improve the confidence of novel SLiM instance prediction allow the identification of SLiM-mediated interactions and interfaces, and finally, allow the consideration of modular architecture of proteins within PPI network analysis.
iELM requires the input of a binary interaction and can be divided into four stages. The first uses 3DID annotation of structural binary complexes to assess if the interaction is mediated by a domain-domain interaction. If so, this putative association is returned and the workflow terminated. If not, the domain identifier is used to identify the SLiM-binding domains . The third stage singles out putative SLiM instances that may bind to the identified domain using the following procedure: SLiMSearch searches the protein sequence of the interacting protein using regular expressions from the ELM resource. This identifies and scores putative instances based on relative local conservation as well as recording the likelihood of the putative SLiM being in a disordered regions using IUPred, and by association their likely accessibility for binding. The overall score for iELM is calculated using a support vector machine (SVM) learning algorithm. The SVM algorithm is a supervised learning method that, based upon a set of input values, assigns weights to each score. These weights can therefore be extracted to determine the importance of each score to the overall score of iELM. The SVM model assigned 25% of the models’ weight to the domain score with similar weights assigned to the RLC scores and the domain length
The final stage of the iELM method is the assessment of the physicochemical likelihood of the interaction using the structural bioinformatics tool, PepSite. PepSite uses spatial position-specific scoring matrices (PSSMs) to find a potential binding site for each amino acid in a peptide. These are then combined to identify the potential SLiM-binding site and a score is calculated that is outputted by iELM with the putative binary complex. PepSite requires a PDB file of a globular domain (see methods for modelling procedures), therefore if there is no PDB structures with a sequence similarity of greater or equal to 30% to the putatively identified SLiM-binding domain then the analysis of physioschemical likelihood by PepSite is not implemented.